GO-based cluster analysis
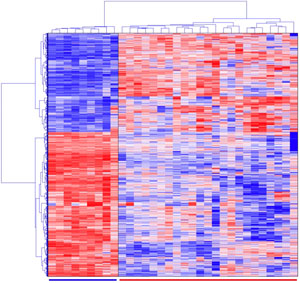 Cluster analysis of microarray gene expression data results in groups of genes expressed similarly under the given experimental conditions, e.g., a drug treatment, a comparison between the normal and disease tissues, or other biological comparisons of interest.
Cluster analysis of microarray gene expression data results in groups of genes expressed similarly under the given experimental conditions, e.g., a drug treatment, a comparison between the normal and disease tissues, or other biological comparisons of interest.
It is well known that similarly expressed (co-expressed) genes tend to share the same or similar function(s) and, in fact, the gene co-expression information is often used for predicting gene functions. Appropriate cluster analysis of microarray gene expression data, therefore, may identify clusters of functionally related genes in genomes. Fuzzy C-Means (FCM, Bezdek 1981) is a popular algorithm that uses a partitioning approach with fuzzy cluster boundaries and fuzzy sets that associate each gene with one or more clusters, representing the fact that when a gene is regulated, it may participate in multiple biological processes. An advantage of FCM is that it converges rapidly, however, like most partitioning clustering algorithms, it depends strongly on the initial parameters and requires estimation of the number of clusters. Cluster validation methods are required to determine the optimal cluster solution. Unfortunately, the clustering model of FCM is a possibility-based one. There is no straightforward statistics-based approach to evaluate a clustering model except the cluster validity indices based on the compactness and separation factors of the fuzzy partitions. However, there are problems with these validity indices; both with scaling the two factors and with over-fit estimates. Recently, we developed fzBLE, a Bayesian based method for fuzzy cluster validation using fuzzy partition, where the possibility model of the fuzzy partition is approximated by a probability model which is used to compute the goodness-of-fit of the model against the data. A common drawback of fzBLE and popular cluster validation methods is the lack of biological knowledge application in cluster validation, resulting in non natural/relevant biological clusters of genes. It yet remains unanswered whether the genes of the same cluster could participate in similar biological processes,molecular function or colocalize in the same cellular compartment, involve in same functional pathways, encode common functional domains and finally induce similar phenotypes. 
Gene Ontology The Gene Ontology (GO) is a hierarchy of biological terms using a controlled vocabulary that includes three independent ontologies for biological process (BP), molecular function (MF) and cellular compartment (CC). Standardized terms known as GO terms describe roles of genes and gene products in any organism. GO terms are related to each other in the form of parent-child relationships. A gene product can have one or more molecular functions, can participate in one or more biological processes, and can be associated with one or more cellular compartments. As a way to share knowledge about functionalities of genes, GO itself does not contain gene products of any organism. Rather, expert curators specialized in different organisms annotate biological roles of gene products using GO annotations. Each GO annotation is assigned with an evidence code that indicates the type of evidences supporting the annotation.
We developed fzGOclust, a combination of FCM and fzBLE algorithms, for cluster analysis of genes set using both the gene expression data and prior biological knowledge. fzGOclust generates fuzzy partition based on a metric of either gene expression levels of GO semantic similarities. Instead of computing the compactness and separation factors using the fuzzy partition, a Bayesian probabilistic model and a log-likelihood estimator is applied to measure the goodness-of-fit of the clustering model. With the use of both the possibilistic model and probabilistic model to represent the data distribution, our method is appropriate for artificial data where the distribution usually follow a standard model, as well as for real datasets, in particular, gene expression data, that lack a standard distribution. We showed that our method outperforms popular cluster indices on gene expression datasets. The method is available online at bioc.tinyray.com. A trial user may use "guest" as username and password to access this utility, running on one CPU core of our high performance computing system (HPC). Thank you for reading this article and love to have your feedback. REFERENCES
- Thanh Le and Katheleen Gardiner (2011) A validation method for fuzzy clustering of gene expression data.
- Thanh Le (2013) A validation method using Gene Ontology for cluster analysis of gene expression data.
- Thanh Le (2013) A Machine Learning approach for Gene Expression analysis and applications.
- Thanh Le (inprep.) A Fuzzy GO-based cluster analysis method for Gene Expression data.